
Se evaluaron los dos métodos para la determinación de nitritos, que solicita la normatividad mexicana, en alimentos infantiles cárnicos con verduras. Se determinó el contenido de nitritos a los alimentos infantiles, materias primas y productos intermedios del proceso de elaboración; en cada corrida analítica se incluyeron un blanco de reactivos y una muestra testigo; además se determinó la sensibilidad, porcentaje de recuperación y precisión de las metodologías. Los resultados en los alimentos infantiles indicaron una importante diferencia en los contenidos de nitritos obtenidos entre las metodologías, debido a la persistente presencia de turbidez en los extractos. Se propusieron diferentes tratamientos físicos para eliminarla, pero únicamente la redujeron; tal turbidez se atribuyó a los hidratos de carbono; las concentraciones de nitritos informadas presentaron una dispersión grande y estuvieron por debajo del límite de cuantificación de ambos métodos, por lo que no es recomendable la aplicación de estas técnicas para alimentos que se sospechan contienen trazas de nitritos.
Palabras clave: Nitritos, método de Griess, alimentos infantiles con carne y vegetales, hidratos de carbono
We evaluated the two methods accepted by the Mexican norm for the determination of nitritesin infant meatbased food with vegetables. We determined the content of nitrites in the infant food, raw materials as well as products from the intermediate stages of production. A reagent blank and a reference sample were included at each analytical run. In addition, we determined the sensitivity, recovery percentage and accuracy of each methodology. Infant food results indicated an important difference in the nitrite content determined under each methodology, due to the persistent presence of turbidity in the extracts. Different treatments were proposed to eliminate the turbidity, but these only managed to reduce it. The turbidity was attributed to carbohydrates which disclosed concentration exhibit a wide dispersion and were below the quantifiable limit under both methodologies; therefore it is not recommended to apply these techniques with food suspected to contain traces of nitrites.
Key words: Nitrites, Griess test, meat-based infant food with vegetables, carbohydrates
Departamento de Ciencia y Tecnología de los Alimentos, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México, D.F., México.
Los nitritos y los nitratos se emplean como aditivos en la fabricación de productos de origen animal y son los responsables del color rosado de la carne curada; contribuyen al sabor e inhibición del crecimiento de Clostridium botulinum cuya toxina puede causar la muerte (1). Su presencia en los alimentos no solo se debe al empleo de sales de curación; por el ciclo biológico del nitrógeno se han encontrado nitratos entre 1 a 10000 mg/kg de peso fresco, en vegetales, diferencia que se deben al suelo, agua y al uso de fertilizantes (2,3). Por otro lado la presencia de nitritos en los vegetales se debe a la reducción del nitrato; reacción que requiere condiciones de pH, enzimas reductoras o iones metálicos; además, ocurre durante el escaldado ó el almacenamiento a temperaturas mayores a las de refrigeración (4).
A manera general se han detectado nitratos y nitritos en alimentos sin procesar como: carne, cereales y pescado, así como en alimentos procesados como: lácteos ó bebidas a base de vegetales; no obstante, los nitritos varían en cada alimento, pero la concentración en productos cárnicos curados no debe ser >156 mg/kg (5), mientras que en alimentos que no se someten a curación se han encontrado concentraciones < 5 mg/kg (2).
En México se comercializan alimentos infantiles (AI) que se etiquetan con nombres de productos cárnicos curados (papilla de jamón con vegetales), lo que hace pensar que fueron elaborados con jamón y son objeto de la normatividad mexicana para ese tipo de cárnicos, aunque el fabricante declara haberlo elaborado con pernil de cerdo que no fue sometido a un proceso de curado.
Debido a lo anterior, es conveniente la correcta aplicación de la regulación por los efectos toxicológicos para el consumidor, formación de Nitrosaminas (1), especialmente cuando los productos van dirigidos a población susceptible, siendo los niños <6 meses los que presentan el mayor riesgo (6).
Entre los métodos analíticos para determinar nitritos en alimentos y agua se encuentran los basados en espectrofotometría, potenciometría, quimioluminiscencia, electroforesis- capilaridad ó HPLC (7-9); en México, la Secretaría de Salud y la industria cárnica, emitieron la norma NOM-122-SSA1-1994, que incluye dos métodos (A y B) basados en la reacción de Griess, debido al bajo costo de reactivos, procedimiento simple, confiabilidad y no requiere instrumentos costosos. Sin embargo, no se ha evaluado su desempeño con AI cárnicos. Un análisis previo en estos productos informó concentraciones < 2 mg/kg con el método B yentre 10 a 20 mg/kg con el método A, diferencia importante, a pesar de que tienen procedimientos similares.
La presente investigación evaluará el desempeño de las metodologías y determinará el origen de las diferencias en los resultados al evaluar nitritos en AI, con los métodos de la normatividad mexicana.
Los AI fueron: Papilla de jamón con vegetales (PJV), Papilla de carne de res con vegetales (PRV) y Papilla de carne de pollo con vegetales (PPV); los cuales fueron proporcionados por el fabricante. La muestra testigo (MT) fue jamón tipo Virginia (JV).
Los métodos, para la determinación de nitritos (A y B), se basan en la diazotización de Griess en la cual se obtiene un colorante azoico por acoplamiento de una sal de diazonio con una amina aromática, que puede ser alfa-naftilamina ó 1-naftilamina en presencia de ácido sulfanílico (Reactivo de Griess). El colorante se detectó a una longitud de onda de 520 nm en un espectrofotómetro Beckman® DU-70.
Ácido sulfanílico, ácido acético, cloruro mercúrico, nitrito de sodio e NaOH de Baker®, 1-naftilamina de Sigma-Aldrich®, sulfato de zinc de Merck® y papeles filtro para retención de partículas de 8, 2.5 y 0.45 μm de diámetro Whatman®.
La diferencia fundamental entre métodos es la extracción, porque la precipitación de proteínas y otros sólidos se realiza con diferentes reactivos.
La extracción del método A se realizó con 1 a 2g de muestra, molida y homogenizada, en un vaso de precipitados de 50 mL, se agregaron 40 mL de agua caliente, se agitó y transfirió a un matraz volumétrico de 250 mL, el vaso se lavo con porciones de agua caliente hasta completar 160 mL. Se colocó el matraz en baño de vapor con agitación por 2 h. Al términose agregaron 5 mL de solución saturada de cloruro mercúrico y se agitó. Se llevó a aforo y se filtró, se tomó una alícuota de 50 mL, en un tubo de Nessler, se agregaron 2 mL del reactivo de Griessse agitó y se dejó reposar durante 20 min para desarrollode la reacción.
Para el método B se realizó con 2 a 3 g de muestra, molida y homogenizada, en un vaso de precipitados de 50 mL, se agregaron 40 mL de agua caliente, se agitó y vacióa un matraz volumétrico de 250 mL, se lavócon porciones de agua caliente hasta completar 160 mL. Se colocó el matraz en baño de vapor por 90 min, se agregaron 10 mL de sulfato de zinc 0.42 M y se agitó. Se agregaron 12 mL de NaOH al 2% se agitó y coloco enbaño de vapor por 10 min. Se enfrió a temperatura ambiente y aforó. Se filtró y tomaron 50 mL en un tubo de Nessler se agregaron 2 mL del reactivo de Griess, se agitó y para desarrollo de la reacción se dejó reposar durante 20 min.
La solución patrón (SP) se obtuvo al diluir 10 mL de una solución que se preparó con 0.5 g de NaNO2 y 1 L de agua libre de nitritos, en 1 L de agua destilada (1mL= 0.005 mg de NaNO2).
Para la curva estándar se consideraron 12 puntos, de 0 a 0.05 mg, que se prepararon al agregar entre 0 a 18 mL de la SPen tubos Nessler, se llevaron a aforó y se agregaron 2 mL del reactivo de Griess, se agitaron y después de 20 min se determinó la absorción del colorante azoico espectrofotométricamente. Los puntos equivalen a concentraciones de 0 a 125 mg/kg de alimento
Los controles para asegurar la calidad de las mediciones, en cada corrida analítica, consistieron en: a. Se preparó la curva de comparación, la cual debió tener una r2≥0.98, b.Se incluyeronla muestra testigo y el blanco de reactivos, c. Las muestras se trabajaron de manera ciega, se codificaron con números aleatorios, y por quintuplicado, d. El funcionamiento del espectrofotómetro se verificó con una celda de holmio.
Se incluyeron dos modificaciones, durante el proceso de filtración, sin afectar el fundamento de las técnicas, con el objeto de eliminar las diferencias entre métodos, tales cambios fueron: a. Previa centrifugación de los extractos a 5500 rpm/5 min y b. Filtración adicional con filtros de 0.45 μm.
Para determinar la sensibilidad, la cual incluye la concentración neta mínima detectable (CMD) y el límite de cuantificación (LoQ), de ambos métodos se empleó la guía Eurachem (10).
Para el porcentaje de recuperación se analizaron soluciones acuosas entre 1 a 75 mg de nitritos/L y AI adicionados, con una concentración conocida de nitritos. Las soluciones se obtuvieroncon la SP y agua desionizada, los resultados se informaron en mg/L, y los AI adicionados se prepararon a partir de 2 g de la papilla a los cualesse agregaron los micro litros (μL) necesarios de SP para obtener 5 concentraciones equidistantes entre 6 a 30 mg/kg, los resultados se informaron en mg/kg; cabe mencionar que en cada corrida analítica se analizó la papilla sin adicionar.
Para la precisión, repetibilidad y reproducibilidad, de ambos métodos se utilizó la ISO-5725-2 (10).
Para determinar la composición de los AI y detectar las posibles interferencias, se llevó a cabo un análisis químico proximal y de fibra soluble e insoluble; de acuerdo a los métodos de normas oficiales mexicanas (NOM) y normas mexicanas (NMX) para extracto etéreo (EE) (11), cenizas (12), humedad (13), proteína (14), fibra cruda (15) y fibra dietética (FD) (16); los hidratos de carbono (HC) se obtuvieron por diferencia.
Los resultados se presentaron con la media, desviación estándar y los coeficientes de variación (CV) para conocer la variación alrededor del valor medio o central, en ambas metodologías.
Para determinar la temperatura y porosidad del papel a emplear; así como, evaluar la diferencia en las concentraciones obtenidas para una misma muestra entre métodos se aplicó una prueba t para muestras relacionadas.
Para determinar como afectanla determinación las variables: porosidad del papel filtro y la temperatura de extracción; el JV se sometió a dos temperaturas de extracción (20 y 92.8°C) en cada método. Los resultados indicaron que esta variable no afectó el contenido de nitritos. Por el método A se obtuvieron concentraciones iguales (p>0.05) en ambas temperaturas y los nitritos promedio estuvieron en 92.88 ± 1.61 mg/kg; en el método B se obtuvo un comportamiento similar yel promedio fue de 75.13 ± 2.65 mg/kg; los CV fueron < 5% y en cada temperatura de extracción la n = 10. Cabe mencionar que el JV, empleado para el desarrollo de cada método fue de diferente lote, lo que explica la diferencia en concentraciones entre métodos. Por lo anterior, la prueba para evaluar el tamaño de poro del papel filtro, se realizó a 92.8°C y se consideraron papeles libres de cenizas y para retención de partículas de 8 μm y 2.5 μm, los resultados fueron diferentes (p< 0.05) por el método A porque los extractos obtenidos con el papel de 8 μm presentaron ligera turbidez, aun cuando se trato de la MT, e informó concentraciones mayores, de 103.01±4.13 mg/kg, no obstante con 2.5 μm se obtuvieron extractos claros y nitritos alrededor de 75.60 mg/kg; el método B no informo diferencias (p<0.05) y la concentración promedio fue de 88.26±2.02 mg/kg; por cada papel la n=10. Debido a lo anterior se decidió emplear una temperatura de 92.8°C para la extracción y papel para retención de partículas de 2.5 μm.
Las materias primas analizadas que se emplean en la elaboración de la PJV,fueron: pernil de cerdo, agua de proceso y almidón modificado de maíz waxy; por el método A se obtuvieron resultados < 3.6 mg/kg y con CV entre 18-46%, el almidón informo la mayor concentración de nitritos seguido del pernil de cerdo; en cambio con el método B se obtuvieron resultados negativos.También se analizaron algunos de sus productos intermedios como: 1. pernil de cerdo/agua, 2. pernil de cerdo/agua/ vegetales, 3. pernil de cerdo/agua/vegetales/espinacas; los resultados fueron menores a 7 mg/kg con el método A, la mayor concentración fue para el producto intermedio 3 y los demásinformaroncontenidos < 1.5 mg/kg con CV >30%, los extractos presentaron turbidez;por el contrario con el método B, no se detectaron nitritos y los extractos fueron claros.
El contenido de nitritos en los AI fueron menores a 9 mg/kg por el método A y la PJV presentóel mayor contenido, 9.05±1.53 mg/kgcon CV= 16.93%; la PRV y PPV informaron concentraciones < 4 mg/kg y con CV>20%; con el método B el único AI que informó una concentración positiva fue la PJV, con 0.98±0.08 mg/kg con CV = 18%.
Los resultados de los tratamientos físicos para eliminar las diferencias entre métodosse presentan en la TABLA 1, en esta se observan concentraciones de nitritos parecidas entre métodos, sin embargo se observa también variación importante.
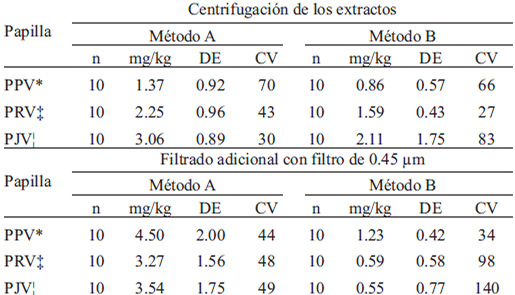
Debido a lo anterior y a las estimaciones de la sensibilidad de los métodos, que se exponen posteriormente, se analizaron los AI cárnicos con untamaño de muestra mayor, para tales corridas analíticas se sometieron al análisis 5g del AI; por el método A no fue posible obtener alícuota suficiente para la reacción; sin embargo, por el método B informaron concentraciones < 1 mg/kg con CV >50%.
De la estimación de la sensibilidad para el método A se obtuvo que la CMD fue de 0.0011mg, que equivale a 2.80 mg/kg, y un LoQ de 0.0037mg (9.33 mg/kg); mientras que, por el método B la CMD fue de 0.0015mg, que equivale de 2.5 a 3.75 mg/kg, y un LoQ de 0.0049mg (8.33 a 12.5 mg/kg); las concentraciones estimadas se calcularon con base al peso de muestra de la normatividad.
Para determinar los porcentajes de recuperación se consideraron las concentraciones entre 1 a 75 mg/kg, porque los AI informaron concentraciones < 10 mg/kg y la MT entre 71 a 75 mg/kg. En ambos métodos, para las concentraciones de 10 a 75 mg/kg los porcentajes se encontraron entre 85 a 118% con CV< 5%, FIGURA 1, no obstante entre 1 a 5 mg/kg se obtuvieron CV> 24% y la recuperación presentó un intervalo mayor, 59 a 127%; debido a los resultados se estableció que las mediciones son mas confiables a partir de 9.7 mg/kg. En todas las corridas analíticas la MT informó similitud (p>0.05) entre métodos y CV< 5%.
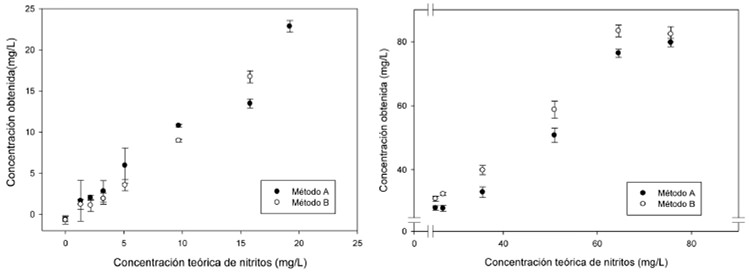
Con el objeto de cuantificar las concentraciones < 9.7 mg/kg con una menor dispersión se llevó a cabo el análisis con 5 y 10 mL de solución de concentración conocida. En el caso de 5 mL se logró cuantificar con CV< 5% y %R entre 80-120 hasta 5 mg/L; sin embargo las menores informaron%R similares pero CV> 15%. Al analizar 10 mL hubo diferencias entre métodos, con el A se cuantificó hasta 2 mg/L con CV< 5%, mientras que con el B fue a partir de 3 mg/L. Cabe mencionar que para estas corridas la curva de calibración se preparó en el intervalo de 0.001 a 0.004 mg de nitrito de sodio y presentó una r2=0.999.
La determinación de los porcentaje de recuperación enlos AI se realizó con las PPV y PRV, se observó que en ambos métodos las papillas sin adicionar y las agregadas con 6 mg/kg de nitritos informaron CV>30%, no obstante, por el método A las concentraciones > 10 mg/kg presentaron CV< 5% y %R entre 80-120%; en cambio el método B tuvo este comportamiento para las concentraciones ≥ 12 mg/kg.
La precisión se realizó con el JV, los resultados para el método A indicaron que 8.94 mg/kg es la diferencia máxima aceptable (DMA) entre las repeticiones del mismo día y una DMA de 9.32 mg/kg entre repeticiones de diferentes días. Para el método B se estimó una DMA de 8.56 mg/kg entre repeticiones del mismo día y una DMA de 10.13 mg/kg entre repeticiones de diferentes días; lo anterior para una concentración de 71.18±1.53 mg/kg en el método A y de 73.15±1.61 mg/kg en el método B.
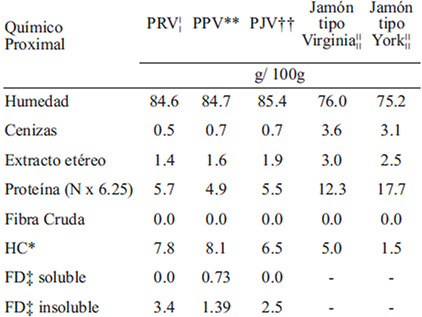
La caracterización se detalla en la TABLA 2; se observa que los AI presentaron de 30 a 40% más de HC, 12% más de humedad, 80% menos cenizas y de 50 a 60% menos proteína y EE, que la MT. Por lo que, la diferencia se ubicó en los componentes relacionados a los HC; pero en el JV, una prueba de lugol confirmó la presencia de HC complejos como almidón y dextrinas, no obstante en el jamón tipo York resultó negativa.
Se realizaron la determinación de las condiciones de temperatura de extracción y porosidad del papel porque no se describen sus características en los métodos de la normatividad mexicana; únicamente se menciona que el papel debe ser libre de nitritos; sin embargo, en los métodos consultados en bibliografía la temperatura de extracción varía entre 20°C a 93°C, temperatura aproximada de ebullición del agua en la Ciudad de México, con tiempos de proceso de extracción de 5 h a las temperaturas bajas y minutos para las altas, dependiendo de la modificación al método (1,7,17).
Por los resultados de nitritos obtenidos en las materias primas y productos intermedios la diferencia entre métodos se pueden atribuir a los HC; así los AI informaron una concentración menor a 7 mg/kg por el método A y menores a 1mg/kg por el método B, las concentraciones mayores se debieron a la PJV; no obstante los tratamientos físicos la disminuyeron, a una concentración menor de 4.5 mg/kg por el método A y menor a 2 mg/kg por el método B pero en ambos casos se obtuvieron concentraciones con gran variación. La concentración para este tipo de productos alimenticios hasta ahora informada se encuentra en 0.05 mg/kg (18).
La turbidez de los extractos obtenidos con el método A se debió al almidón modificado, se ha informado que presenta opacidad en solución en todo el espectro visible, 400 a 700 nm, con porcientos de transmitancia (%T) a 650 nm entre 5.20 a 20.9 porque depende de las modificaciones y sustituciones realizadas al almidón de maíz waxy (19-21), debido al principio óptico de la espectrofotometría es primordial que no exista turbidez porque interfiere con las lecturas (22); el almidón modificado empleado en estos AI informa el menor %T (21), además con esta metodología únicamente se desnaturalizan las proteínas, por el contrario con el método B se obtuvieron extractos claros lo que se atribuye a la adición de NaOH, el cual modifica el pH del medio y ocasiona que las proteínas pierdan su capacidad de asociación con los HC; también, se ha informado que provoca ligera ionización del almidón y debido a las fuerzas electrostáticas de los grupos hidroxilo, aniónicos, se evita la asociación de las cadenas del almidón por repulsión (19).
Por otro lado, debido a los resultados en la determinación de la sensibilidad no sería recomendable la aplicación de estas metodologías para evaluar productos que se sospecha contienen concentraciones más bajas al LoQ, pues estas concentraciones presentaran una variación importante. Una estrategia para cuantificarlas es incrementar la cantidad de muestra, pero es viable para muestras que presentan extractos claros, en concordancia en métodos espectrofotométricos para verduras se emplean 25g de muestra diluida a 200mL para concentraciones altas y 20g diluidos a 50mL para bajas, con lo cual la CMD fue de 0.2 mg/kg (23), esta posibilidad se descartó para el método A. No obstante con el método B al incrementarse el tamaño de muestra se detectaron concentraciones por debajo de la CMD, pero coincidentemente presentaron gran variación.
La precisión de los métodos se comprobó a la concentración informada por la MT; sin embargo, a las concentraciones que presentaron los AI no fue posible estimarla porque estuvieron por debajo del LoQ y CMD.
De la recuperación de los métodos, con los AI, los porcentajes fueron aceptables y de acuerdo a los estándares, es decir, entre 80 a 120%; sin embargo para concentraciones menores a 10 mg/kg los resultados presentaron este mismo comportamiento, no obstante el CV se incrementó; además, a manera general, se encontraron diferencias en las concentraciones informadas por cada método, por el método B fueron más cercanas a la concentración teórica, situación contraria en el método A lo que se puede atribuir a la persistente opacidad de los extractos. En últimas investigaciones, se ha corroborado y determinado que la presencia de ácido ascórbico interfiere en la reacción del compuesto azo, lo que puede resultar en una menor detección de nitritos (24). Además un pH ácido, menor a 5, afecta la estabilidad del nitrito, mientras que un pH básico y alrededor de 8.86 sólo se extrae el 68% del nitrito presente en la muestra, por lo que se ha sugerido un ajuste de pH entre 6 a 8 pero puede ocasionar la precipitación de las proteínas, lo que restringe la matriz alimenticia a analizar, (25).
Los resultados de la presente investigación fueron decisivos en la modificación de la NOM-122-SSA1-1994 (5), misma que fue derogada y sustituida por la norma oficial mexicana NOM-213-SSA1-2002, en esta última se hace referencia a la implantación de controles de análisis para asegurar la calidad de las determinaciones; así como también se cambió la sal diazo: alfa-naftilamina (NAFTILAMINA 1) por N-1-naftiletilendiamina (NED), ya que representa menor riesgo para la salud del analista.
Sin embargo los AI o sus materias primas no son regulados aún en México, en cuanto al contenido de nitratos o nitritos; en la Unión Europea, para regular el contenido de nitratos y nitritos en alimentos procesados y no procesados, se han emitido dos leyes, Reglamento 194/97 y la Directiva 95/2/EC y la FDA dentro del Código de Regulación Alimentaria, estableció un máximo de 200 mg de nitrito/kg de alimento, como agente conservador, pero en la etiqueta debe portar la leyenda “No debe ingerirse por infantes” ya que para esta población se recomienda una ingestión no mayor a los 10 mg/ kg.
La Ingestión Diaria Aceptable (IDA) de nitratos, informada por el comité conjunto de la FAO/OMS 2002, es de 0 a 3.7 mg/kg peso corporal. Puesto que la toxicidad de los nitratos proviene de su conversión en nitritos, deberá tenerse en cuenta también la IDA de nitritos que se estableció de 0 a 0.07 mg/kg de peso corporal.
Los métodospara la determinación de nitritos de la normatividad mexicana son aplicables para productos alimenticios que contengan concentraciones de nitritos > 9.5 mg/kg, para tales concentraciones las metodologías estudiadas informan CV menores a 5% lo cual es deseable. En estos métodos la principal causa de obtención de falsos positivos por el método A es la turbidez, por lo que es importante considerar que para productos que contengan almidones modificados es recomendable aplicar el método B, con el que se logra la clarificación. Es importante hacer énfasis que estas metodologías no son aplicables para los AI cárnicos con vegetales, además se deberá evitar utilizar nombres de productos cárnicos curados para nombrar productos alimenticios que no los contienen y por último se sugiere que se modifique la NOM-213-SSA1-2002 para que se informe los LoQ de los métodos, con lo que se evitara se analicen alimentos con una concentración > 9.5 mg/kg.
Recibido: 27-04-2012
Aceptado: 11-10-2012